
Copyright © W. J. Heitler (2019)
Synapses
Synapses are the communication nodes of the nervous system. They are the sites where one neuron passes on information to another neuron, or to an effector organ such as a muscle. So synapses enable the nervous system to make an animal actually do something - to behave. However, synapses not only transmit information, they also enable neurons to integrate that information and make decisions based on it (to spike or not to spike). Essentially, it is synapses that enable the brain to think. So synapses are important.
This chapter describes simulations concerning the post-synaptic electrical events that occur at:
- Chemical synapses
- Electrical synapses.
It also has sections on:
There is a tutorial on pre-synaptic inhibition in the Network section.
Chemical Synapses
At a chemical synapse, information is transmitted between neurons in the form of a chemical signal. Specifically, depolarization in the output region of a pre-synaptic neurone opens voltage-dependent calcium channels, and the resulting inflow of calcium triggers the exocytosis of vesicles containing the chemical signal, the neurotransmitter. This diffuses across the synaptic cleft and interacts with receptors on the post-synaptic membrane. It is this interaction which causes electrical events in the post-synaptic neuron.
In a spiking chemical synapses only a spike in the pre-synaptic neuron generates enough depolarization to cause transmitter release. In a non-spiking chemical synapses, sub-threshold pre-synaptic depolarization can cause transmitter release on its own, without the occurrence of a spike.
Excitation and Inhibition
- Load and Start the parameter file Excite Inhibit.
Three neurons are visible as yellow blobs in the Setup view. A line ending in a blue diamond runs from N1 to N3; this represents a synaptic connexion. A similar line runs from N2 to N3. The diamond shape shows that the synapses are of the spiking chemicalNon-spiking chemical synapses are shown as rounded-rectangles, and electrical synapses are shown as squares. type, and the labels a and b indicate that they have different propertiesNote that the synapse labels a and b are arbitrary. They indicate that they are different types of synapses with different properties, but they have no meaning outside the program. Synapse properties are set using the Synapses menu command.. N1 and N2 are both stimulated with positive current (square boxes 1 and 2) and spikeThe neurons in these simulations are modelled using an integrate-and-fire algorithm. This means that the spikes are just digital events that trigger synaptic output, they are not implementing the HH equations. This is much faster, but it means that the appearance of the spikes in the Results view is just cosmetic. in response, generating post-synaptic potentials (PSPs) in N3.
In the Results view we see the membrane potentials of the 3 neurons, plus a stimulus monitor under each one.
Question: One of the synapses generates an excitatory post-synaptic potential (EPSP) in N3 while the other generates an inhibitory post-synaptic potential (IPSP). Which is which?
-
- Apply and remove \(\alpha\)- bungarotoxinα-Bungarotoxin is derived from snake venom (kraits of the genus Bungarus) and is a specific blocker of nicotinic acetylcholine receptors. and bicucullineBicuculline blocks GABAA receptors in turn by checking and unchecking the appropriate box in the Drugs group of the Experimental Control panel.
- Run-on-change and Auto clear have been pre-selected in the Results view for your convenience.
Does the outcome confirm your ideas regarding EPSPs and IPSPs?
- Apply and then remove TTX.
These synapses require spikes in the pre-synaptic neuron to trigger transmitter release. This is not always the case, as we will see shortly.
- Uncheck the Auto clear box in the Results view.
- Set the Amplitude of stimulus 3 to 0.25 nA.
We have now injected a stimulus into N3 to raise its membrane potential during the time of the PSPs. You should see that the EPSPs have got smaller in terms of their base–to-peak height, while the IPSPs have got bigger.
- Now set the Amplitude to -0.25 nA (just put a minus sign in front of the number).
Now the EPSPs have got bigger. The IPSPs, however, have reversed their direction. Instead of going negative from their base potential, they go positive!
Question: Why do the PSPs change in base-to-peak amplitude, and why do the IPSPs reverse polarity with hyperpolarization? You can assume that the conductance change underlying the PSPs is not affected by the post-synaptic membrane potential. Hint: Think about equilibrium potentials and driving force.
Integration
In a real nervous system, an individual post-synaptic neuron may receive input from manyPerhaps up to 1000 different pre-synaptic neurons, some of which is excitatory and some inhibitory.
- Load and Start the parameter file Integration.
Again we have a post-synaptic neuron (N1, blue) receiving excitatory and inhibitory synaptic input, but this time there are 6 excitatory neurons (e2 - e7, red) and 6 inhibitory neurons (i8 - i13, green) pre-synaptic to N1. The pre-synaptic neurons each receive a tonic, randomly-varying stimulus current which makes them spike at random intervals, and thus deliver EPSPs and IPSPs to the post-synaptic neuron. Due to the random nature of the stimulus current, the spike pattern will vary with each simulation run. The strength of each synapse has itself been randomized (see the Connections: Randomize menu command), meaning that each synaptic connection varies in the strength of the input it makes to N1.
- Check the Spike frequency graph box in the Display mode group of the Results view to show the Spike Frequency dialog. This is non-modal and can be kept visible while running simulations.
- Move the Spike Frequency dialog box to a position where you can see both it and the Results themselves.
- Click Start a few times to run several simulations.
The Spike Frequency dialog shows a bar graph of the cumulative spike frequency of each neuron. The post-synaptic neuron, N1, is the left-most column in the chart. N1 receives an about equal balance of excitation and inhibition, and spikes at about the same frequency as the pre-synaptic neurons themselves.
- Check the bicuculline box in the Drugs group of the Experimental Control panel to block the inhibitory (GABAergic) input to the post-synaptic neuron.
Note that without inhibition the frequency of post-synaptic spikes increases considerably; it is now about twice the frequency of the pre-synaptic neurons. This is apparent both from the density of spikes in the top trace in the Results view, and from the height of the bar in the N1 column of the Spike Frequency dialog.
- Remove the bicuculline and apply \(\alpha\)-bungarotoxin.
Now the post-synaptic neuron N1 never spikes, because it only receives inhibitory synaptic input.
Take-home message: The post-synaptic neuron integrates its synaptic inputs (essentially sums them together), and if the resulting membrane potential rises above threshold it generates a spike signal, which may then be passed on to other neurons. If the post-synaptic neuron does not spike, no signal is passed on. The propagation of a signal thus depends on the balance of excitation and inhibition in each neuron in a circuit.
Voltage Clamp
Ligand-gated (synaptic) channels can be examined under voltage clamp just like voltage-dependent channels. (A general description of the voltage clamp technique was given previously, and you could review it if you need a reminder.) The advantage of the voltage clamp technique is that it can directly reveal the currents underlying the PSPs, which in turn can allow the conductance to be calculated.
- Load and Start the parameter file Excite Inhibit VC.
Despite the filename, the simulation starts in normal current clamp mode, so you see the voltage response in the post-synaptic neuron. As before, two pre-synaptic neurons are activatedThe activation times and synapse properties can be examined and specified by checking the Properties box in the Synapses group in the Setup view. and in this case generate a single spike each, but these are not visibleThis is because the simulation is using the Advanced HH module, which only shows one neuron. This module is needed because it implements the voltage clamp protocol, which is not available in the Network module.. They cause an EPSP followed by an IPSP in the post-synaptic neuron. Note: to keep things simple, in this simulation the post-synaptic neuron has no voltage-dependent channels (it is a non-spiking neuron). The only ion channels in the membrane are the leakage channels which generate the resting potential, and the ion channels underlying the two PSPs.
- Apply and remove the drugs \(\alpha\)BT and bicuculline in turn.
- The results suggest that the EPSP is mediated by nicotinic cholinergic receptors, and the IPSP by GABAA receptors.
- In the Setup view, select Voltage as the Clamp mode (click Yes to clear the screen when prompted).
- Click Start.
The upper trace shows the membrane potential, which is completely flat because the clamp potential is set the same as the holding potential, which is itself set to the unclamped resting potential of -60 mV.
The middle trace shows the total current generated by the voltage clamp, which mirrors the ionic current across the membrane. The initial clamp current is 0. This is because the membrane is clamped at the resting potential, and no clamp current is needed to maintain this voltage. However, 5 ms after the start of the run, the excitatory synapse is activated, and there is a downward deflection in the current trace reflecting the inward current flowing through the cholinergic receptor channels. The downward deflection is because the clamp has to inject negative current to prevent the positive current flowing through the open ion channels from shifting the membrane potential in a positive direction. A little later the inhibitory synapse is activated, and there is an upwards deflection in the clamp current trace counteracting the outward current flowing through the open GABA receptor channels.
The lower trace shows the conductance of the two types of channel generating the PSPs (EPSP red, IPSP green). Remember that these cannot be recorded directly in an experiment - they are calculated during the simulation from the underlying data.
One feature that is immediately obvious is that the current pulses (and underlying conductance changes) are much briefer than voltage change in the membrane potential seen in the initial experiment before the voltage clamp was applied. This is because the capacitance of the membrane slows down the voltage response to a current pulse (see the Basic RC Properties section if you want a reminder about this).
Question: Why are the current profiles for the two PSPs (middle trace) so different from each other in amplitude and direction, when the underlying conductance changes (bottom trace) are identical?
- Uncheck the Auto clear box in the Results view.
- Click the up arrow of the Clamp 1 Potential spin button to change the clamp to -45 mV. A new sweep occurs because Options: Run on change has been pre-selected.
- Repeatedly click the up arrow until the clamp potential is +15 mV.
As soon as a depolarizing clamp step is applied, the current trace shows a step change upwards. This reflects the leakage current - the current needed to keep the membrane potential elevated above its natural resting potential. A little later, the synapses are activated and there is additional clamp current needed to counteract the current flowing through the open receptor channels.
- Without clearing the screen, expand the timebase by clicking the Results toolbar expand button (
 ) twice. This should make it easier to see the current data for the first PSP (the EPSP).
) twice. This should make it easier to see the current data for the first PSP (the EPSP).
Task: Find the equilibrium (reversal) potential for the EPSP. Hint: The clamp current during the PSP is made up of a combination of the leakage current and the synaptic current; these need to be separated to find the PSP equilibrium potential. This can be done by constructing separate I/V plots for the components. The following protocol reflects one commonly found in published research papers.
-
- First, outside of Neurosim, open a suitable graphing program such as Excel.
- Within Neurosim, check the Measure box in the Results view to open the Measure dialog.
- The starting position of the red measure cursor should be within the clamp step, but before the synapse is activated (i.e. before the EPSC ). The current in this position is just leakage current, there is no EPSC. This is what we want, so there is no need to move the cursor.
- Click the Measure button in the Measure dialog.
- The values from the 6 experimental sweeps will appear in the Measure display.
- The clamp potential is in the Voltage (mV) column, and the clamp current is in the I Tot (nA) column.
- Click Copy, and paste the measurements into Excel.
- We now have the data needed to construct an I/V curve for the leakage current.
- Click Clear in the Measurement dialog (not in the Results view).
- In the Results view, move the red measure cursor so that it aligns with the peak of the conductance shown in the bottom trace (time about 5.2 ms).
- Repeat the measurements as above. Be careful not to overwrite the first set of measurements when pasting into Excel.
- We now have the data to construct an I/V curve for the leakage plus synaptic current.
- In Excel, construct a single graph showing the I/V plots for both sets of data on the same axes.
The graph below shows the expected outcome of the task:
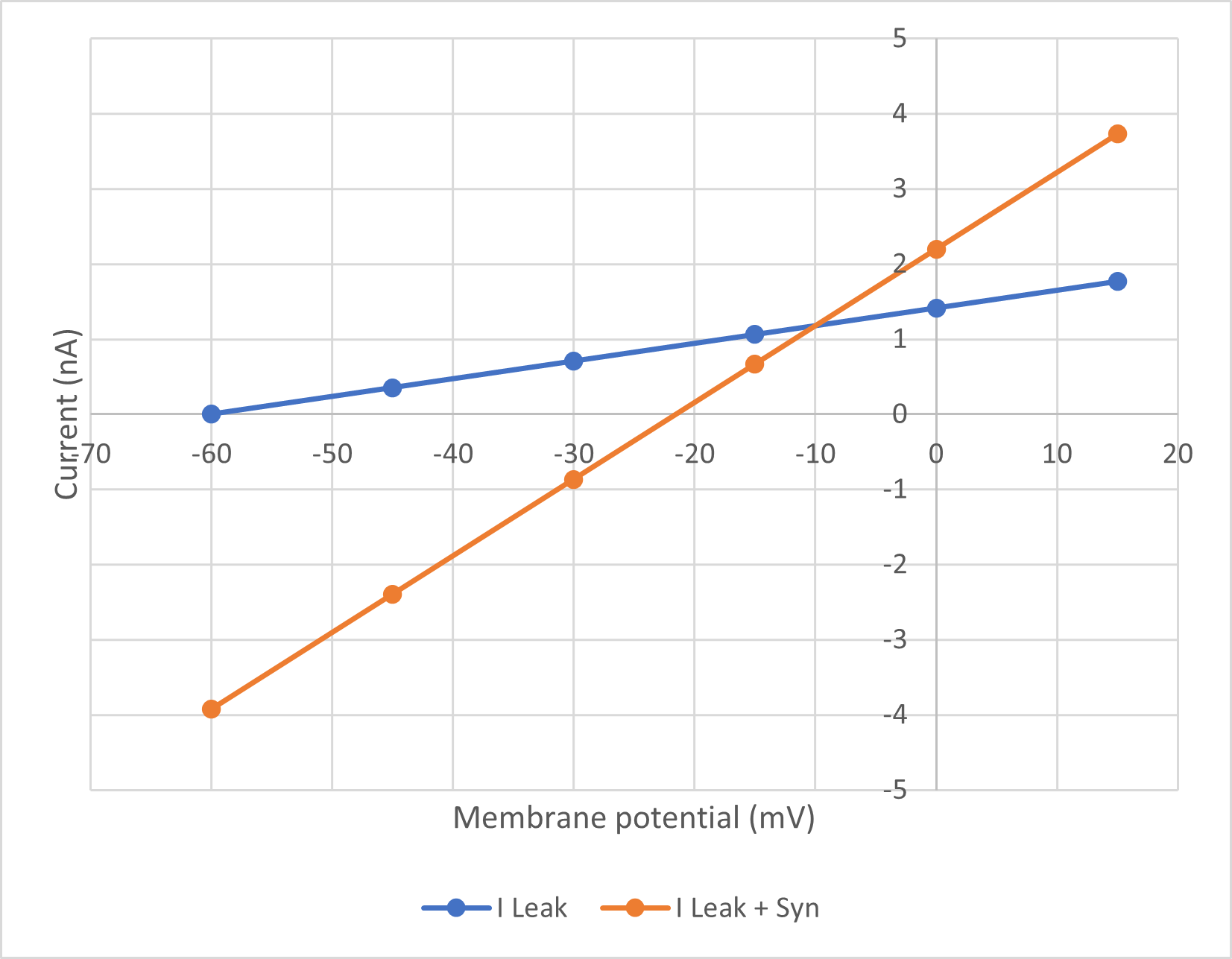
The blue line is the leakage current on its own (IL), the orange line is the combined leakage and synaptic current (IL + IS). Where the lines intersect the two are equal, so the synaptic current is zero (if IL = IL + IS, then IS = 0). The voltage at which this occurs is thus the equilibrium potentialThis follows from the driving force equation I = g(Em - Eeq). We can assume that g is non-zero, because there is synaptic current at voltages on either side of the intersect (IL ≠ IL + IS). So if I is zero but g is non-zero, Em - Eeq must be zero, meaning Em = Eeq. for the synaptic current.
- Click the Results toolbar Show all button (
 ) to bring the current data for the IPSP back into view.
) to bring the current data for the IPSP back into view. - Click Clear, and then click Start to produce a single trace.
- Repeatedly click the down arrow for the Clamp 1 potential spin button until the clamp potential is -90 mV.
Question: What is the equilibrium potential for the IPSP?
Hint: You could use the same approach as for the EPSC, but for this simulation there is a simpler way. You can just note the clamp potential at which the current trace is a flat line - i.e. the clamp current at the peak of the inhibitory conductance is equal to the leakage current. This is possible because the equilibrium potential just happens to exactly match one of the voltage clamp steps!
Summation and Facilitation
- Load and Start the parameter file Facilitation Summation.
The Setup view shows is a simple circuit in which N1 makes an excitatory synapseThe synapse label is again a, but the properties are different from the previous a. As stated earlier, the label IDs are arbitrary onto N2. N1 is driven to spike at quite high frequency by a train of brief stimuli (look at the stimulus 1 parameters). The EPSPs increase in peak amplitude as the train progresses.
Question: Do the EPSPs show summation, facilitation, or both?
Remember that summation is a post-synaptic phenomenon, when the voltage change produced by closely-spaced EPSPs sum with each other, whereas facilitation is a pre-synaptic phenomenon, in which successive spikes release greater amounts of transmitter.
- Change the mean interval of stimulus 1 to 50 ms and click Start.
The stimuli to N1 now occur at lower frequency, and the EPSPs in N2 are more separated in time. Does this affect your answer to the previous question?
Depolarising and Silent IPSPs
- Load and Start the parameter file Depolarizing IPSP.
At first site, the synapse looks like it is generating EPSPs, but in fact, the response is inhibitory despite the fact that it is depolarising.
- Set the amplitude of stimulus 2 to +0.2 nA.
- Click Start.
Note that the PSPs that looked like EPSPs now look like IPSPs!
- Increase the amplitude of stimulus 2 to +0.3 nA.
- Click Start.
The stimulus is now big enough to generate spikes in the post-synaptic neuron, and you can see that the IPSPs interrupt the spike train, thus showing that even though they are depolarising when the post-synaptic neuron is at rest, they are none-the-less really inhibitory.
This type of depolarising inhibition is quite common, especially in pre-synaptic inhibition. It usually arises from an increased chloride conductance when the chloride equilibrium potential is depolarised relative to the resting potential, but hyperpolarized relative to spike threshold.
Hyperpolarizing IPSPs inhibit neurons through two mechanisms. First they simply take the membrane potential further from threshold, so it takes more excitation to generate a spike. But secondly, they have a “shunting” effect. This means that the increase in conductance that generates them will tend to short-circuit any excitatory currents that are generated by nearby concurrent EPSPs.
Depolarizing IPSPs lack the first mechanism, but they certainly include the second. They may also have an additional property that can make them even more effective. The small depolarization that they generate can activate voltage-dependent potassium channels, and start to inactivate sodium channels. They thus effectively render the patch of membrane where they occur somewhat refractory, thus reducing the probability of it generating a spike. This is similar in principle to spike threshold accommodation, which you may have seen earlier.
Take-home message: A PSP may generate a depolarizing potential when activated at the resting potential of the post-synaptic neuron, but that does not of itself tell you whether it is excitatory or inhibitory.
Absolutely nothing seems to happen in the post-synaptic cell, despite the pre-synaptic spikes.
- Set the amplitude of stimulus 2 to +0.1 nA.
- Click Start.
Now IPSPs are clearly apparent!
The explanation here is that the synapse generates an increase in chloride conductance in the post-synaptic neuron (N2), but the chloride equilibrium potential is the same as the resting membrane potential. Therefore the driving force is zero and there is no current, and hence no voltage change. However, as soon as you perturb the membrane potential to take it away from the chloride equilibrium potential, the PSPs are revealed.
Non-Spiking Synapses
Transmitter release from the pre-synaptic terminal is triggered by an inflow of calcium through voltage-dependent calcium channels. In many neurons, these channels have a high threshold, and only a spike invading the pre-synaptic terminal can depolarize it enough to open the channels and cause transmitter release. However, in some neurons the calcium channels have a low threshold, and transmitter can be released just by depolarizing the terminal without generating a spike. Indeed, in some neurons transmitter is released even at resting potential, and its release can be modulated by any change in potential in the pre-synaptic neuron.
- Load and Start the parameter file Non-Spiking Synapse.
Nothing appears to happen.
N1 connects to N2 through a non-spiking chemical excitatory synapse. The properties are similar to those of the nicotinic acetylcholine synapse, but in this case transmitter release from the pre-synaptic neuron does not require spikes. Instead, transmitter is released in a graded manner, with increased pre-synaptic depolarisation leading to increased release of transmitter, up to a saturation level.
- Increase the amplitude of stimulus 1 in steps of 0.1 nA until N1 starts to spike, and observe the post-synaptic response. You can just repeatedly click the up-arrow of the Amplitude spin button.
- Apply TTX to the preparation, and note that pre-synaptic spikes are not required for the synapse to operate.
- Now remove the TTX and set the current back to 0.
- Clear the screen.
- Inject increasingly negative levels of current into N1 by clicking the down-arrow of the Amplitude spin button until the level reaches -0.5 nA.
Note that for the first few stimuli, both N1 and N2 hyperpolarize. But then, while N1 continues to hyperpolarize, N2 does not.
Question: Why does N2 hyperpolarize at all? Why does the N2 hyperpolarization not increase beyond a certain level as you increase the hyperpolarization of N1? Hint: think about turning off a dripping tap.
Non-spiking synapses are not obscure oddities. In fact, if you are reading this tutorial, you are using them right now. The rods and cones of the vertebrate retina do not generate spikes, but release transmitter at a level determined by their membrane potential. They are depolarized in the dark and continuously release transmitter, and this release is reduced by light-activated hyperpolarization.
Voltage-Dependent Synapses (NMDA-type)
Some post-synaptic receptors at chemical synapses have a voltage-dependent conductance. The classic example is the NMDAN-methyl-D-aspartate-receptor. The natural agonist for this is glutamateThe receptor also requires glycine or D-serine to bind to it for full activation., but glutamate can also activate a variety of other receptors. A highly specific artificial agonist is NMDA, from which the receptor derives its name.
The voltage-dependency results from magnesium block. At the normal resting potential, magnesium ions, which are naturally present in the extracellular fluid, partially block the channel at resting potential. A magnesium ion literally plugs the channel, fitting into the channel lumen from the extracellular side. This means that even if an agonist has bound to the channel and the structural channel "gate" is open, the channel still has a low conductance. As the post-synaptic neuron depolarisesI.e. the intracellular space becomes more positive., the positively-charged magnesium ions are increasingly expelled from the channel, thus relieving the block. This means that as the neuron depolarizes, the overall conductance of activated NMDA channels increases. The depolarization needed to remove the magnesium will reduce the driving force on the normal carrier ions (sodium and calcium), but usually not enough to significantly reduce their current.
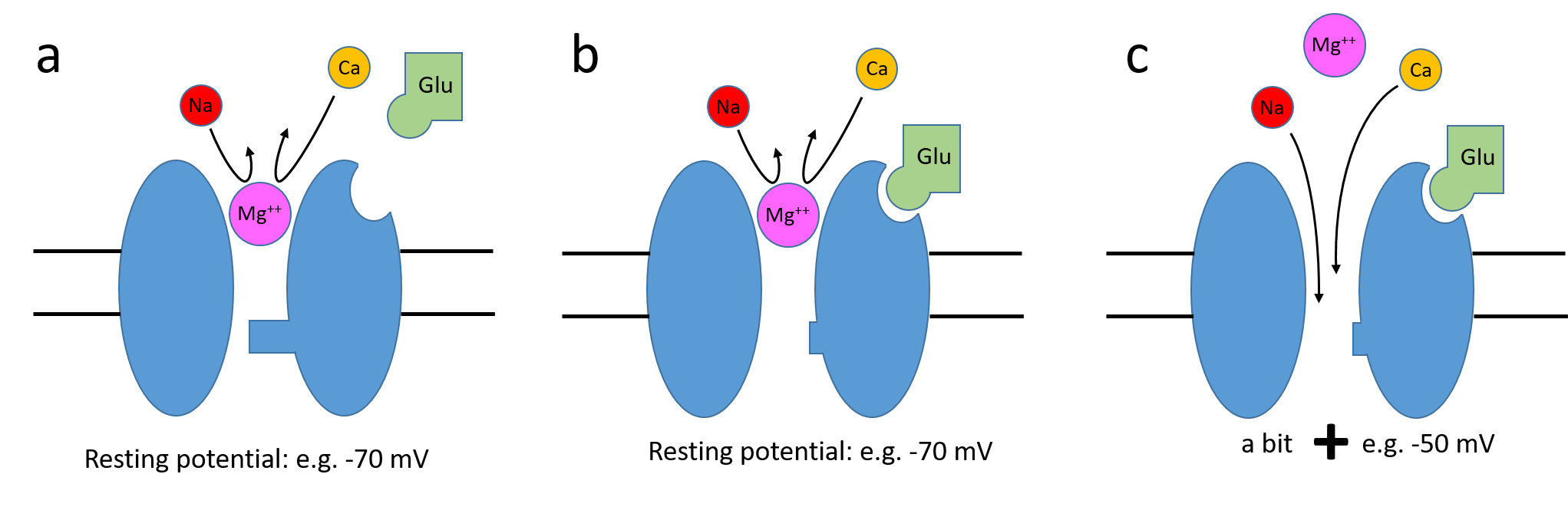
This looks just like the excitatory synapses that we have seen earlier, although it is quite small. For clarity, this neuron has no voltage-dependent channels, so it's response is entirely driven by the synapse E that is activated 10 ms after the start of the simulation (the activation time is set in the synapses Properties which we will see later). The Synapses list shows that E is an NMDA-type synapse.
- Without clearing the screen, click the up-arrow of the spin button for the amplitude of stimulus 1, to inject 0.1 nA of positive current into the post-synaptic neuron, timed to bracket the EPSP.
- Repeat twice more, to bring to current to 0.3 nA.
Note that the size of the EPSP has increased as the neuron depolarizes! This is not what is expected for a normal excitatory PSP. Depolarizing the post-synaptic neuron takes its membrane potential closer to the equilibrium potential for an excitatory synapse, and thus the driving force decreases and the size of the EPSP should decreaseIf it were a depolarizing IPSP then its size should also decrease with depolarization, even more than for an EPSP.. But in this case it increases.
The reason is that as the cell is depolarized by the stimulus, magnesium is progressively expelled from the receptor channel, and hence when the channel is activated by binding the ligand (glutamate in nature, NMDA in the lab), it achieves a higher conductance than at resting potential, when the magnesium block is stronger.
The biological significance of this is that NMDA-receptor mediated responses have a tendency for self-reinforcement; as the synapse becomes more active (perhaps due to increased frequency of pre-synaptic spiking, or by spatial summation with other excitatory synapses), the effectiveness of transmission is increased in a non-linear fashion, so that the post-synaptic cell rapidly depolarises to a higher potential level.
- Set the Stimulus 1 current back to 0.
- Clear the screen.
- Set the right-hand timebase scale to 250 ms.
- Check the Props box in the Synapses list to open the Synapse Properties dialog.
Make sure that the new dialog does not obscure the Results view.
The Properties dialog is quite complicated, but for now just note that Type E is selected in the list at the top left, indicating that we are looking at the properties of Type E synapses. The description shows that this is an NMDA type. The Active box is checked, indicating that it is active, and the Latency setting is 10 ms, indicating that it will be activated 10 ms after the start of the simulation.
- Check the Repeat box for the type E synapse (this is the only Repeat box that is enabled).
Note that the repeat Interval is 6 ms, and the Count is 5.
As soon as you check the box, the Results view updates showing that the synapse is activated 5 times, at 6 ms intervals. The EPSP shows standard facilitation, but there is nothing much interesting about the response.
- In the Synapse Properties dialog, set the type E repeat Count to 50.
Now the synapse is activated repeatedly for a long duration. At first, it shows standard facilitation, but after while there is a sudden increase in the rate at which the EPSP size increases. This is the positive feedback effect of removing the magnesium block the channel.
- Just for comparison, uncheck the Make voltage dependent box in the Voltage-dependent properties group.
Unchecking the box effectively completely removes the magnesium block. In a real experiment this could perhaps by achieved by bathing the preparation in an extracellular fluid containing no magnesium, and hence preventing magnesium block. The EPSP is much bigger from the outset, but the facilitation shows a standard progression without the pronounced non-linearity visible when the block is enabled.
At this point you should have 3 sweeps superimposed.
- Use the highlight sweep facility to in the Results view to compare the 3 sweeps in turn.
The first sweep shows a brief set of 5 EPSPs, the second an extended set of EPSPs showing non-linear facilitation due to the magnesium block, and the third an extended set of EPSPs showing standard facilitation which achieve approximately the same final amplitude as the second, but which start from a much larger amplitude.
NMDA I/V Curve
We can use the voltage clamp technique to look at the I/V curve for an NMDA-receptor mediated synapse: i.e. the relationship between post-synaptic synaptic current and post-synaptic voltage.
- Reload the file NMDA to return to the starting conditions.
- Click Voltage in the Stimulating: Clamp mode group of the Setup view to switch to voltage clamp mode.
- Click Start.
The upper trace shows the clamp membrane potential. It starts off at the holding potential of -60 mV, which is the resting potential of the neuron. It then briefly steps down to -90 mV, during which time the synapse is activated. The second trace shows the current flowing through the NMDA receptor channel (leakage current has been removed). However, there is no visible deflection in this trace, indicating that 0 or virtually 0 current is flowing through the channel. This is because the channel is almost completely blocked by magnesium at this membrane potential. The conductance (bottom) trace is also essentially flat at 0 (but remember that conductance cannot be measured directly in a real experiment - it is calculated from the driving force equation).
- Repeatedly click the up arrow of the Clamp 1 Potential spin button, until the clamp potential reaches +20 mV, looking at the current trace after each click. Do not clear the Results between each run.
A visible deflection should appear in the current trace after the second click when the clamp potential is -70 mV. This indicates that the magnesium block is starting to be relieved.
Task: Construct an I/V plot showing how synaptic current varies with clamp potential, using the following procedure:
-
- Check the Measure box in the Results view to show the Measure dialog.
- Drag the red measure cursor to line up with the peaks of the conductance in the bottom trace.
- The peaks should all occur at the same relative time in the sweeps, which is about 10.5 ms.
- Click Measure in the dialog to make a simultaneous measurement of all the traces in all the sweeps, at the time of the cursor location.
- Click Plot in the dialog to open the XY Scatter dialog.
- Select Voltage from the X axis drop-down list.
- Select I Syn (synaptic current) from the Y axis drop-down list.
You should now see a J-shaped curve which is very similar to the I/V curve produced by voltage-dependent sodium channels. This is not a coincidence - both channels have the region of negative slope conductance characteristic of regenerative processes that can cause a rapid, spike-like depolarization. Look back at the sodium experiment if you want a reminder about this.
Question: What is the approximate equilibrium potential of the NMDA response? Supplementary question: Do you need to worry that your answer could be affected if the receptor is partially blocked by magnesium?
Note: The NMDA receptor is a mixed cation channel, and the charge-carrying ions are a mixture of sodium, potassium and calcium. So the effective equilibrium potential for the channel will not be the Nernst potential for any one of these specific ion, but rather a weighted-average of the 3 Nernst potentials.
You know the synaptic current at each potential from the voltage clamp data, and hopefully you now know the equilibrium potential from the I/V curve, so you could now calculate the conductance at each potential using the driving force equation. However, Neurosim does this for you, and displays the results in the conductance trace. Furthermore, you will have picked up the value of each peak conductance when you measured the current.
Task: Construct a plot showing how the conductance of the activated NMDA channel varies with membrane potential.
This is extremely simple - just select G syn from the Y axis drop down list in the XY Scatter dialog (G is the standard symbol for conductance).
Question: At what membrane potential is the magnesium blockade fully relieved - i.e. what is the membrane potential that expels all the magnesium ions from all the NMDA receptor channels?
Hebbian Synapses
An early theory of learning (devised by a man called Hebb, hence the name) was that learning occurred as a result of increases in synaptic strength that resulted from the “conjoint firing” of pre- and post-synaptic neurons (Hebb, 1949). Hebbian synapses are synapses that exhibit this phenomenon: their strength is increased when the pre- and post-synaptic neurons are active together. In its simplest form this means that when an excitatory Hebbian synaptic input is “successful” in eliciting a spike in the post-synaptic neuron, the synapse gets strengthened (neurons that "fire together, wire together", Hebb, 1949). This means that the synapse is likely to be more effective the next time it is activated. Conversely, if the Hebbian excitatory synaptic input is not followed by a spike in the receiving neuron, then the connexion does not get strengthened, or can even get weakened.
N1 spikes at constant low frequency (due to synaptic input from an unspecified neuron) and puts small EPSPs onto N2. In the middle of the sweep N2 is induced to spike by an injected current pulse (this could be a synaptic input from some other neuron). After the N2 spike, the synaptic input from N1 is much enhanced - thus when the N1-N2 connexion was "successful" (due to help from the stimulus) it got stronger. Note that the synapse in the Setup view is colour-coded according to its strength.
- When you are clear about the phenomenon, select Scroll box in the Trigger group on the Results view.
- Clear the screen.
- You will probably have to slow the simulation down (depending on your computer speed), so select a Slow-down factor of 6 from the main toolbar. (The best factor depends on the speed of your computer - you can adjust it while the simulation runs.)
- Click Start.
Notice that as time passes after the Hebbian effect, the enhanced response diminishes back to its starting state. The synapse has “forgotten”.
- When the EPSP has decreased substantially but with the simulation still running, click the Stim button within the Manual stimulus group at the bottom-left of the Results View.
If the manual stimulus generates a spike in N2, it causes another enhancement of the EPSP.
- Click Stim repeatedly, until the EPSP has grown enough in amplitude to cause the post-synaptic neuron to spike without the additional manual stimulus.
At this stage, the post-synaptic neuron spikes in response to each EPSP and so the synapse receives reinforcement on each input and thus maintains the enhanced response, even with no further injected pulses. The synapse has “remembered” its reinforcement.
- Click Pause to give yourself some time.
- Change the Manual stimulus amplitude to -2 (minus 2), and the duration to 500.,
- Click Continue to resume the simulation.
- Click Stim again, repeating if necessary. By blocking spikes in the post-synaptic neuron with negative current, we can make the synapse “forget” its enhancement.
The post-synaptic spike is an essential feature to trigger Hebbian synaptic enhancement, and yet the enhancement itself may well involve the increase in transmitter release from the pre-synaptic neuron. This implies the once-heretical notion of a retrograde transmitter. This means that the post-synaptic neuron releases some substance when its activity level increases that has an effect back across the synapse specifically onto the active pre-synaptic terminal.
There is as yet no direct evidence that any real synapse displays Hebbian properties which are as simple as those in this simulation. However, Hebbian-type mechanisms may well underlie some of the important forms of learning which occur in the nervous system, such as long-term potentiation (LTP) and classical conditioning.
Quantal Release
At the heart of our modern understanding of chemical synapses is the concept of quantal release – the notion that transmitter is released in fixed-size packages, now known to be vesicles, from a pool of available vesicles, and that the number of vesicles released can vary. Katz and co-workers (del Castillo & Katz, 1954) established this in a series of classic papers that match the Hodgkin-Huxley papers in their influence, and which also led to a share in the Nobel prize for Katz.
- Load and Start the parameter file Quantal Release.
This simulates a classic experiment. A neuron makes a connection to another neuron via a cholinergic synapse. The pre-synaptic neuron is stimulated repeatedly at a fixed frequency of 50 Hz, and the post-synaptic response is recorded. The Setup and Results views only show the post-synaptic neuron (the simulation is running in the module Advanced HH), and the pre-synaptic neuron is not shownThe pre-synaptic stimulus frequency and the synaptic properties are set in the Synapse Properties dialog box. If you wish, you can access this by checking the Properties box in the Synapses frame of the Setup view, but you don’t need to for this activity..
There is obviously some variation in the size of the EPSPs, but it is hard to see any obvious pattern.
- Reduce the external calcium concentration to 1 mM.
- The screen should clear and run a new experiment (because Results view Auto clear and the Options: Run on change menu have been pre-selected).
The size of the EPSPs reduces substantially. This is because transmitter release is calcium-dependent.
- Further reduce the external calcium concentration to 0.1 mM.
- Click Start a few times to get a good impression of the different sizes of EPSP.
You might notice two things:
First, there are occasional “gaps” in the response. The pre-synaptic neuron is spiking at a constant frequency (you have to just believe that – it is part of the simulation), but quite frequently it fails to elicit a response in the post-synaptic neuron.
Second: the size of the EPSP varies in relatively fixed steps, although because there is some noiseNoise, in this context, means random fluctuations in the signal that carry no useful information. in the recording (added intentionally in the simulation) the steps are not always absolutely equal.
The latter property can be seen more clearly as follows:
- Click Clear.
- Uncheck the Auto clear box in the Results view.
- Select Triggered from the Results view Trigger mode group.
- Set the sweep time base maximum (the bottom right axis scale) to 10 ms
- Click Start (the external calcium concentration should still be 0.1 mM).
You are now seeing an expanded view of the individual PSPs that occur over the same time period (400 ms), but the PSPs are all lined up and superimposed. This is what is meant by “triggered”. The display sweep is synchronized to the time when the membrane potential crosses a user-specified Trig level (-59 mV in this case), and this happens on the rising front of the EPSPs. The display also captures some Pre-trig signal (set at 2 ms) so you can see the baseline activity just before the EPSP. This is standard display methodology frequently used in real experiments.
- The results are probabilistic, so click Start several times, clearing the screen between runs, to see the variety of results.
It should now be obvious that the size of the EPSPs is not continuously graded. Instead, EPSPs cluster into discrete size classes:
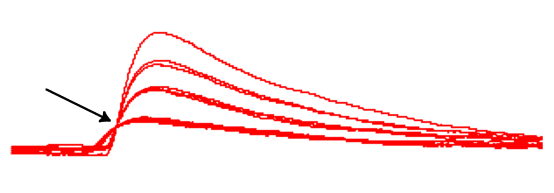
- With the screen clear, increase the external calcium concentration to 0.3 mM
You should now see rather more steps in the EPSP amplitude.
- Again with the screen clear, increase the external calcium concentration to 1 mM
The size of the EPSP is still clearly changing in steps, but there are (probably) none of the really small EPSPs visible. You may also start to see some slowly declining traces that are obviously not EPSPs. These are caused by trigger-crossing due to noise on the falling edge of the EPSP and can be ignored.
The data can be interpreted as follows. There is a pool of releasable vesicles in the pre-synaptic terminal, and with each pre-synaptic spike a random number of these are released, with the number fluctuating around some average value (the average quantal content). In reduced calcium, this average is reduced.
At the frog neuromuscular junction, where these experiments were first carried out, at normal levels of extracellular calcium, the average quantal content (average number of vesicles released per stimulus) is high, so transmission is very reliable. However, in synapses in the central nervous system, the average quantal content may be only 1 or 2, so pre-synaptic spikes frequently produce no post-synaptic response at all, because no vesicles are released.
Quantal Size Distribution
You should only read this is you are interested in the statistical properties of quantal release.
Assume that the total number of quanta available for release is n, and that the probability of any one quantum being released in response to a single pre-synaptic spike is p. This means that the average quantal content of an EPSP, m, will be equal to np. The probability P(k) of k quanta in total being released for a particular stimulus is given by the binomial distribution:
\begin{equation} P(k) = \frac{n!}{k!(n-k)!}p^k(1-p)^{n-k} \label{eq:eqBinomial} \end{equation}The problem with this formulation is that although we can measure m experimentally (the average size of the PSPs divided by the minimum size), we cannot measure n or p, and so if we detect a change in m we do not know whether the effect has been mediated by changing the probability of release (p), or the number of quanta available for release (n).
One of the useful characteristics of the binomial distribution, is that if m is low compared to n, the distribution simplifies to a Poisson distribution
\begin{equation} P(k) = \frac{m^k e^{-m}}{k!} \label{eq:eqPoisson} \end{equation}This is still a bit of a mouthful, but the key point is that there are no unmeasurable quantities in the equation, and the releasable pool variable n doesn’t come into it. So any change in m that may occur means that p has changed, rather than n.
In order to use the simple Poisson equation, we need to be sure that quantal release follows a Poisson, rather than a binomial, distribution i.e. that m (the average number of quanta released) is small compared to n (the total number of quanta available for release). Happily, there is a fairly simple test we can apply.
When we stimulate the pre-synaptic neuron it sometimes fails to release transmitter at all, i.e. the quantal content is 0. We can measure the probability of this happening as simply the fraction of the total number of stimuli which fail to evoke EPSPs, i.e. the number of failures, N0, divided by the number of trials N. From the Poisson distribution above we know that:
\begin{equation*} P(0)=\frac{N_0}{N} = \frac{m^0e^{-m}}{0!} \end{equation*}m0 and 0! are both 1, so this resolves to
\begin{equation*} N_0 = Ne^{-m} \end{equation*}and hence
\begin{equation} m = \ln\frac{N}{N_0} \label{eq:eqQuantalM} \end{equation}It was this sort of analysis that led to the conclusion that facilitation was usually due to an increase in the mean quantal content (m) of transmission.
Electrical Synapses
Electrical synapses are mediated by gap junctions that form an electrically-conducting cytoplasmic bridge from one neuron to another. This means that current can flow intracellularly between neurons, so that if one neuron spikes, the depolarizing current entering that neuron spreads to the other neuron, which consequently receives an electrical EPSP. This has two important consequences. First, electrical synapses are faster than chemical synapses because they do not involve the intermediary chemical signal. This means that electrical synapses are often found in neural circuits mediating behaviours where speed is essential, such as escape behaviours. Second, most electrical synapses can “share” both excitation and inhibition between pools of neurons, thus tending to synchronize them. So electrical synapses are often found in pools of neurons that perform similar functions and which often spike together, such as the motor unit pools innervating a particular muscle.
- Load and Start the parameter file Electrical Synapse.
N1 and N2 are connected by an electrical synapse (the square synapse symbol in the Setup view). The synapse icon might suggest that N1 is anatomically pre-synaptic to N2, but that is just for consistency in the Setup layout, it does not represent the physiology.
First a pulse of positive current is injected into N1, where it produces a subthreshold depolarization. This spreads through the electrical synapse into N2, where it produces a somewhat smaller depolarization. So in this situation, N1 is presynaptic to N2. Second, a pulse of positive current is injected into N2, where it also produces a sub-threshold depolarization, which spreads to N1. So in this situation N2 is presynaptic to N1. The terms pre- and post-synaptic are thus operationally defined at this type of synapse - whichever is the source of the current is the pre-synaptic neuron, and this can change on a moment-by-moment basis.
The standard way to quantify the strength of an electrical synapse is to measure the coupling coefficient (Cf), which is simply the ratio of the post-synaptic voltage change (Vpost) to the pre-synaptic voltage change (Vpre). This relates to the synaptic and membrane conductance as follows:
\begin{equation} Cf = \frac{\Delta V_{post}}{\Delta V_{pre}} = \frac{g_s}{(g_s + g_{post})} \label{eq:eqCouplingCoefficient} \end{equation}where gs is the synaptic conductance and gpost is the membrane conductance (reciprocal input resistance) of the post-synaptic neuron. (Note that the coupling coefficient is thus independent of the pre-synaptic membrane conductance. This conductance relationship can be shown by applying Ohm’s law, but I will not prove that here.)
Task: Measure the coupling coefficient for both stimuli, using the appropriate voltage values from the two neurons. To make it (marginally) more interesting, the two current pulses do not have quite the same amplitude. Hint: don't forget that it is the change in membrane potential that we are interested in.
The electrical synapse conductanceThis can be viewed in the Electrical Synapse Types dialog, which is accessed through the Synapses: Electrical menu command. is 10 nS, and the membrane conductanceThis can be viewed in the Neuron Properties dialog, which is accessed by right-clicking a neuron in the Setup view. of each neuron is 15.7080 nS. Does your measured coupling coefficient fit with these values?
NEM (n-ethylmaleimide) is a drug that blocksAt least in some types of tissue. It is not absolutely reliable. gap junctions.
- Clear the screen, apply NEM, and click Start.
Two changes are apparent. There is no longer a PSP in either neuron in response to the current injected into the other. That’s because the drug worked. Also, N1 starts to spike. [The simulation is using integrate-and-fire dynamics, so the spikes are not full HH types].
Question: Why does N1 spike when you add NEM? (And why doesn’t N2?) This is a slightly trick question in that it relies on the exact parameters of this simulation, so it is not a general principle. But it is worth considering because the answer helps firm-up understanding of what is happening at these synapses.
- Remove the NEM.
- Set the amplitude of Stimulus 1 to -0.5 nA (just reverse its polarity).
- Clear the screen and click Start.
Take-home message: Simple electrical synapses are bidirectional and non-polarized. Current of either polarity will spread from either neuron to the other. This is in obvious contrast to chemical synapses, which normally mediate one-way information transfer from the pre- to the post-synaptic neuron.
Frequency Matters
Because of cell membrane capacitance, the coupling coefficient of an electrical synapse depends on the frequency of the signal being transmitted.
- Load and Start the parameter file Frequency Matters.
This is a bit complicated, but can be understood by going through it carefully.
There are two identical cell pairs, N1 + N2 (red, blue) and N3 + N4 (green, orange). The members of each pair are connected to each other by identical electrical synapses. N1, the top neuron on the left in the Setup view (red), is stimulated by a standard square pulse of current (stimulus 1). N3, the top neuron on the right (green), is stimulated by current that oscillates in a sine wave with a frequency of 50 Hz (stimulus 2).
In the Results view, the bottom axis shows the stimulating current applied to the two neurons. Note that the peak current for the oscillating (ACAC: alternating current. This is the term used when current varies with time. ) stimulus (green) is higher than that for the stable (DCDC: direct current. This is the term used when current is stable over time.) stimulus (red). The top axis shows the voltage responses of the two pre-synaptic neurons. They both reach the same peak amplitude (apart from the first cycle of the sine wave, which is larger due to the need for settling time).
Question: Why are there differences between the peaks of the AC and DC stimuli, when they both produce the same peak voltage response?
Hint: remember that due to its RCRC: resistor-capacitor properties the membrane acts like a low-pass filter.
The membrane potentials of the post-synaptic neurons, N2 (blue) and N4 (orange), are shown superimposed in the middle axis. Note that the voltage change in the post-synaptic neuron receiving oscillating input through the synapse (N4) is smaller than that receiving constant input (N2), even though the peak voltages in the pre-synaptic neurons are the same.
- With data displayed on the screen, edit the time base axes to set the start time of the display to 125 ms and the end time to 175 ms. This will zoom in the view on the central portion.
- Activate a vertical cursor by clicking the Add cursor button (
 ) in the Results toolbar (or use the View: Cursors: Vertical: Add menu command).
) in the Results toolbar (or use the View: Cursors: Vertical: Add menu command). - Position the cursor over the left-hand peak of the sine-wave stimulus (green trace, bottom axis). This should be at about 135 ms.
Note that the voltage response in the pre-synaptic cell (green trace, top axis) is delayed relative to the stimulus. The peak membrane voltage occurs after the peak stimulus current. This is a pre-synaptic phenomenon, and nothing to do with the electrical synapse.
- Now move the cursor rightwards to the peak of the oscillating pre-synaptic membrane potential (green trace, top axis) which occurs at about 137.7 ms.
Note that the peak of the post-synaptic response (orange trace, middle axis) occurs after the peak of the pre-synaptic signal, indicating a further phase delay introduced by the electrical synapse.
Take-home message: When transmitting a signal, electrical synapses attenuate and delay high frequency signals more than they do low frequency signals.
Size Matters
Although most electrical synapses are bidirectional and non-polarized, the relative size of electrically coupled neurons can have a strong functional influence.
- Load the parameter file Size Matters and look at the Setup view.
N1 and N2 are connected to each other through an electrical synapse (square blue synapse), but N1 is bigger than N2 (100 µm in diameter compared to 50 µm). Both neurons receive an experimental current stimulus (square boxes 1 and 2).
- Click Start.
Both N1 and N2 spikeThese spikes are generated using the HH equations. and their spikes are identical in size and shape. They each generate an electrical EPSP in their partner neuron, but these are very different in amplitude. The N2 EPSP (received by the small neuron from the large neurons, seen early in the blue trace, middle axis) is substantially bigger than the N1 EPSP (received by the large neuron from the small neuron, seen late in the red trace, top axis).
- To make it easier to see the difference, zoom in by setting the top and bottom voltage scales to -44 and -74 mV for both the top and middle axes. The spikes go off screen, but the difference in EPSP sizes is more obvious.
Take-home message: If a big neuron and a small neuron are electrically coupled, a spike in the big neuron makes a bigger EPSP in the small neuron than a spike in the small neuron makes in the big neuron. The synapse is effectively polarized.
The reason can be seen from the equation \eqref{eq:eqCouplingCoefficient} defining the coupling coefficient. A small post-synaptic neuron will have a lower membrane conductance (gpost) than a big neuron due to its reduced surface area, so the denominator in the equation is reduced and the coupling coefficient is bigger.
Rectification
Most electrical synapses are electrically symmetrical: they act as a simple low-resistance path between the neurons. However, some electrical synapses are rectifying and act like an diode. The conductance (inverse resistance) of such a synapse depends on the trans-junctional potential, i.e. the difference in membrane potential between the two neurons. What this means in practice is that positive current will pass in one directionRemember that positive current flowing from A to B can be carried by positively charged ions such as K+ flowing from A to B, but it can just as easily be carried by negatively charged ions such as Cl- flowing from B to A. more easily than negative current, while negative current will pass more easily than positive current in the other. If the trans-junctional potential is such that the synapse is in the high conducting state, the synapses is said to be forward biased. If it is in the low conductance state, it is reverse biased.
Two neurons are connected by a rectifying electrical synapse. The direction of rectification is from N1 to N2 (shown as a diode image in the Setup view). Four stimuli (the square boxes in the Setup view) are applied in sequence.
The first stimulus is positive current that depolarizes N1. It forward-biases the synapse, which moves into a high-conductance state, and the current flows from N1 into N2 and depolarizes it.
The second stimulus is negative current that hyperpolarizes N1. The synapse is reverse-biased and does not move into a high conductance state. Only a little current flows into N2, and the N2 response is weak. Note that the N1 response is actually bigger, because it is not “losing” so much current into N2.
The third stimulus is positive current that depolarizes N2. However, this is on the “wrong” side of the rectifier, so it reverse-biases the synapse, and little current flows into N1 and the N1 response is weak.
The fourth stimulus is negative current that hyperpolarizes N2. This forward-biases the synapse, because it is the relative membrane potentials (i.e. the voltage across the synapse itself, from one intracellular compartment to the other) that determines the bias state of the rectifier. Consequently, the synapse moves into a high conductance state, and negative current flows from N2 into N1, and hyperpolarizes it.
Crayfish
Rectifying electrical synapses are relatively rare, but have been found in both vertebrates and invertebrates. In fact, the very first physiological evidence for the existence of electrical synapses came from investigating the properties of a rectifying electrical synapse that occurs in the neural circuit controlling the escape tail-flip of the crayfish (Furshpan and Potter, 1959). Several other discoveries of general importance have come from studying this circuit (including the one in the next tutorial), so I will spend some time describing it.
Crayfish have two types of escape tail-flip; a backward flip elicited when the attack is from the front, and a forward flip (actually, more of an “up-and-over” somersault, but forward gives the concept) elicited when the attack is from the rear (see Sillar, et al., 2016 chapter 7 for review).
Crayfish escape tail-flip. A juvenile crayfish (on the left) is attacked from the rear by a dragonfly larva. It escapes with a forward somersault! (The sequence is shown twice - the second time in slow motion.)
Video courtesy of Prof Jens Herberholz, University of Maryland.
Both types of tail-flip are caused by rapid flexionThe abdominal cavity is almost entirely filled with flexor muscle, and it is this that makes crayfish and lobster and scampi a food item that many people (and other predators) enjoy. of the abdomen, but there is a crucial difference between the two. For backwards escape the flexion is uniform across the whole abdomen (blue in the upper panel of the figure below), resulting in a jet of water being projected forward with the reaction force thus propelling the animal backward (blue arrow). For forwards escape the flexion is centred on the thoracic-abdominal joint, with the posterior abdomen remaining straight (red in the lower panel of the figure below), resulting in a jet of water being projected downwards and slightly backwards and the reaction force driving the animal upwards and forwards (red arrow).
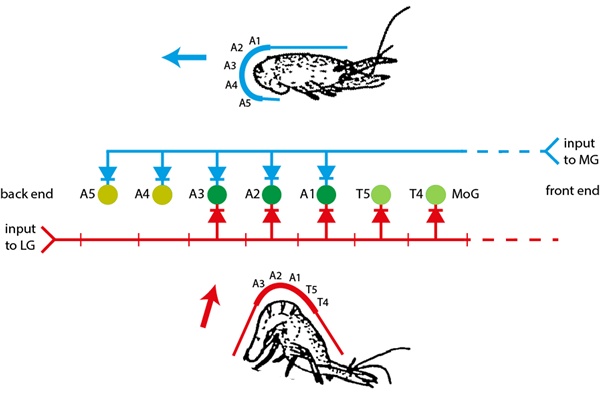
The tail flexion itself is driven by a chain of very large segmental motorneurons known as the motor giant (MoG) neurons. MoGs receive excitatory synaptic input from two bilateral pairs of giant commandCommand neuron is the name given to a single neuron that can trigger an entire behaviour when it spikes. There has been much debate about whether such neurons really exist, mainly centred around arguments about what constitutes an “entire” behaviour, but I’m going to use the term anyway. neurons, the medial giant (MG) and lateral giant (LG). The MGEach bilateral MG is a single interneuron that extends the length of the animal from the brain to the end of the tail. is only activated by a threat from the front and makes input to all the abdominal MoGs but not the thoracic MoGs. The LGThe LGs are actually a set of repeating segmental interneurons. However, each LG is coupled to its segmental neighbour by a very powerful non-rectifying electrical synapse, so the chain of LGs acts as if it were a single neuron extending from the posterior tail to the brain, just like the MG but in the opposite direction. is only activated by a threat from the rear and makes input to the MoGs in the anterior abdomen and posterior thorax, but not those in the posterior abdomen. It is this difference in MoG activation pattern that mediates the crucial difference in flexion curvature.
Both the command neurons drive the MoGs through powerful rectifying electrical synapses (the diode symbols in the figure). These are so strong that a spike in the pre-synaptic command neuron (MG or LG) always elicits a spike in the post-synaptic MoG.
Why does the rectification matter? It matters because the MoGs in the anterior abdomen (A1-3, dark green in the figure) get input from both MG and LG. If the synapse were not rectifying, then a spike in the MG would activate the MoG, and the spike in the MoG could then pass through and activate the LG, and vice versa. Thus a spike in either type of command neuron would activate the other type, and the resulting tail flexion would be some sort of combined hybrid of the two which would be highly unlikely to mediate effective escape in either direction. Rectification prevents this disaster, thus maintaining the speed and reliability of an electrical connection, while ensuring that the transmission is one-way like a chemical synapse.
This is a simple simulation of the flexion command elements of the circuitThe circuit is actually bilaterally symmetrical, but only one side is shown in the simulation. Furthermore, the circuit does not include the elements controlling the tail-fan, which are located in the terminal ganglion, nor any of the circuit anterior to the 4th thoracic segment. controlling the tail-flip that demonstrates the importance of rectification.
There are 3 rows of elements in the Setup view. The MG (top row, blue) is modelled as a 7-compartment single neuron, where each compartment is located within a posterior thoracic or abdominal segment. It makes powerful rectifying electrical synapses (square cyan boxes b) to the MoGs (second row), but only in the abdominal segments (A1-5; dark green, beige). The LG (bottom row, red) is modelled as 7 separate neurons connected in a chain by powerful non-rectifying electrical synapses (square cyan boxes a). The LG also makes powerful rectifying electrical synapses to the MoGs, but only in the posterior thoracic and anterior abdominal segments (T4, 5, A1-3; light green, dark green). The simulation thus replicates the connectivity shown in the figure above.
- Click Start.
The MG is activated by sensory inputTypically sudden mechanosensory or visual input. at the head end, represented by the electrical stimulus in the square box 1. The MG spike propagates along its axon (the first and last compartments are shown in the top axis of the Results, blue) and drives spikes in the abdominal MoGs (A2 and A5 are shown in the middle axis of the Results, dark green and beige) but not in the thoracic MoGs (T4, pale green). The LG remains silent (bottom axis, red).
- Change the Target neuron in the Stimulus group of the Experimental Control panel from 1 to 8, to apply the stimulusNow the stimulus represents sudden mechanosensory input to the tail, such as a shock wave from the predatory strike seen in the video above. to the LG in the tail.
- The Results are cleared and a new sweep occurs because Auto clear and Run on change are pre-selected.
The LG is activated (first and last segmental neurons are shown in the bottom axis, red) and drives spikes in the anterior abdominal and thoracic MoGs (dark and pale green) but not the posterior abdominal MoGs (beige).
The pattern of activation can be seen more clearly by colour-coding the activity in the Setup view.
- Clear the Results view.
- Select the Neuron: Colours: Colour from voltage menu command.
- All the neurons in the Setup view show as dark blue because nothing is active.
- Click Start.
- If necessary, set a Slow down factor greater than 0 to see the pattern of activity more clearly.
A spike propagates anteriorly in the LG, and as it does so it activates the MoGs in the anterior abdomen and posterior thorax. The MG, and the posterior abdominal MoGs, remain silent. The spikes in the 3 mid-chain MoGs (in the anterior abdomen) do not feed through to activate the MG, because the synapse connecting the MoG to the MG is rectifying (in the MG → MoG direction), and so the spike in the MoG reverse-biases the rectifier which therefore remains in a low conductance state.
What happens if we remove the rectification?
- Select the Synapses: Electrical menu command to open the Electrical Synapse Type dialog box.
- If possible, move the dialog so that it does not obscure the Setup or Results view.
- Select b: rectifying synapse in the synapse list.
- Note that the Make rectifying box is checked for this synapse type.
- Uncheck the Make rectifying box.
As soon as you make the change a new sweep is started, and now all the MoGs are activated, and so is the other command neuron, the MG. This is apparent in the colour coding of the elements in the Setup view, and also in the traces in the Results view. There are 3 spikes in the MoG traces (middle axis) showing that the MoGs in the anterior, middle and posterior of the chain (T4, A2, A5) are all active. This would probablyI say "probably" because this has never actually been tested experimentally (there is currently no known drug or genetic manipulation that can switch off rectification like we can in the simulation) but it seems highly unlikely. not produce an effective escape.
Question: The spikes in the anterior and posterior compartments of the MG (top axis, blue) occur virtually simultaneously, while those in the LG (middle axis, red) show posterior-to-anterior conduction delay. Why is that?
- Change the Target neuron in the Stimulus group of the Experimental Control panel back to 1, to re-apply the stimulus to the MG in the head.
Again, all the MoGs are activated, along with the LG.
- Finally, re-check the Make rectifying box in the Synapse dialog.
- You should now return to the normal response to MG activation, which is for the LG to remain silent, and for just the abdominal MoGs to spike.
- Click OK to close the Synapse dialog.
Hopefully, this simulation has demonstrated why rectification is necessary for electrical synapses in some circuits.
Electrical Synaptic Current
You can visualize the current through the electrical synapses as follows:
- Reload the file Crayfish to return to the starting conditions.
- Click the Traces button to open the Trace and Axis Setup dialog.
- Enter:
- Axis 1 : Neuron 4
- Axis 2: Neuron 18
- Axis 3: Neuron 11.
- This will show just the MG, MoG and LG responses in the middle of the chain.
- Check the Show single neuron details box at the bottom-right to expand the dialog.
- Note that the Neuron ID (top left of expansion) is pre-set to 18 (the MoG).
- Check the Current Show box (towards the bottom left of the expansion).
- Note that the Electrical synapse box is pre-checked, as is the Show 0 box.
- Click OK to close the dialog.
- Click the Expand timebase toolbar button () to zoom in on the LG-MoG interaction.
- Click Start.
The bottom axis in the Results view shows the total current entering and/or leaving the MoG through the electrical synapses.
The current profile has a simple monophasic waveform - a pulse of positive current enters the MoG from the pre-synaptic spike in MG, and this depolarizes the MoG and induces it to spike.
- Reopen the Electrical Synapse Types dialog.
- Select b: rectifying synapse in the synapse list.
- Uncheck the Make rectifying box.
- Click Start.
The MoG current now has a tri-phasic waveform. First, a pulse of current enters the MoG (positive) from the pre-synaptic spike in MG, and as before, this is what causes the MoG to spike. Second, a pulse of current leaves the MoG (negative) as the MoG spike depolarizes the LG. Third, another pulse of current enters the MoG (positive) from the post-synaptic spike in LG. This "backwash" does not cause significant depolarization in the MoG because the latter is highly refractory at this time.
Question: Why does the backwash current have a double-hump shape? Hint: remember that current through a electrical synapse depends on the voltage difference between the two connected neurons.
Neuromodulation
What is Neuromodulation?
With neuromodulation, there is a neuron that releases a chemical signal, usually through exocytosisBut not always: gasotransmitters like nitric oxide (NO) are neuromodulators that can diffuse across the plasma membrane and do not require vesicles for release, nor cell-surface receptors for detection. from dense core vesicles, but in contrast to a normal chemical synapse the releasing neuron does not make anatomical contact with any specific post-synaptic neuron with receptors for that signalHowever, the neuromodulator may be co-released with a standard transmitter that does operate through a normal chemical synapse, e.g. nociceptors release the standard transmitter glutamate and the neuromodulator Substance P, although the latter only occurs when the nociceptor spikes at high frequency, signalling intense pain.. Instead, the signal enters extracellular space and diffusesNeuromodulators can also be transported by mass fluid flow through the circulatory system. throughout a volume of tissue (which can be up to the whole organism) where it can affect any neuron that contains receptors for the signal. The response is thus usually relatively slow compared to a standard chemical synapse. Furthermore, most neuromodulators work through metabotropic mechanismsAn exception is the serotonin 5HT3 receptor sub-type, which is a cation-selective excitatory ionotropic receptor. (i.e. through second-messenger biochemical cascades), and so their effects can be quite long lasting. A given neuron in a network may have receptors for multiple neuromodulators, and its response will dependent on the balance between the different signals. Thus in many ways a neuromodulator signal is like a hormone, but the key difference from a standard hormone is that neuromodulators are, by definition, released by neurons, and so their release depends on neural circuit activity. Furthermore, the receptors are located in neurons (although not necessarily exclusively so), and so their effect is to modulate neural circuit activity.
Typically, a neuromodulator can alter (increase or decrease) the conductance/current flow through a synaptic or voltage-dependent channel, but only if that channel is activated in the normal fashion. Thus a synaptic neuromodulator may have no effect on a synapse unless and until the synapse is activated by its normal transmitter (or a suitable artificial agonist), but then it may reduce or increase the synaptic current relative to the strength it would have had in the absence of the neuromodulator. Similarly, a neuromodulator of a voltage-dependent channel may alter the conductance of that channel, but only if the trans-membrane voltage is within the range that normally activates the channel. This relatively simple form of conductance/current modulation is the only mechanism of neuromodulation currently implemented in Neurosim.
In real systems, neuromodulation can have more complex effects such as altering the dynamics of the response, and not just the level of response (see. e.g. Nadim & Bucher, 2014, for a review). Thus a synaptic neuromodulator may have frequency-dependent effects by changing the rate of facilitation or decrement of the synapse (or even switching from one to the other), while not affecting the post-synaptic response to a single pre-synaptic spike at all. Or for voltage-dependent channels, the neuromodulator may alter the range of voltage activation, or the time course of activation/inactivation, while not altering the steady-state value of conductance. In fact, almost any property of any channel can be affected by neuromodulation, and there is physiological evidence for a very wide variety of such effects. However, these more complex effects cannot be directly simulated in Neurosim (although of course the user can usually alter the appropriate parameters manually to see the effect).
Synaptic Neuromodulation
Post-synaptic neuromodulation
In some systems synaptic neuromodulation works by altering the conductance of an activated ionotropic receptor.
- Load and Start the file GABA modulation.
A pre-synaptic neuron (N1) makes an inhibitory GABAergic synapse onto a post-synaptic neuron (N2). N1 is induced to spike by a depolarizing stimulus, and N2 consequently receives a barage of IPSPs.
There is a third neuron (N3) which is not connected to the other two neurons directly, but which releases the neuromodulator serotonin (5HT) when it spikes (the orange circle connected to N3 in the Setup view represents this output). However, at this stage N3 is silent, and the extracellular neuromodulator concentration (lower trace) remains at zero throughout.
- Click the up spin button for the amplitude of stimulus 2 (which is pre-selected in this simulation).
- Because the Run on change option is pre-selected, a new simulation sweep occurs.
Now N3 fires 4 spikes at 80 ms intervals, and the extracellular neuromodulator concentration builds up to a peak of about 2 unitsNeuromodulator concentration units are arbitrary in Neurosim., before declining once the releasing neuron ceases spiking.
The first N3 spike has no apparent effect because the GABAergic synapse is not active at that time. However, once the synapse is activated, the post-synaptic response in N2 is considerably reduced due to the existing non-zero neuromodulator concentration. The reduction is not uniform, but co-varies with the neuromodulator concentration shown in the bottom trace. Thus towards the end of the barage of IPSPs their amplitude increases back towards the control level, due to clearance of the neuromodulator once N3 stops spiking.
Serotonin is a naturally-occuring neuromodulator, but it is also commercially-available as a pharmacological agent that can be applied exogenously during an experiment.
- Check the 5HT box in the Drugs/Light frame of the Setup view.
Now serotonin has been pre-applied as a drug at concentration 1. The effect sums with the endogenously-released serotonin and the overall modulatory down-regulation is enhanced.
Note: The quantitative aspects of the simulation above are plausible, but the values are not based on specific experimental evidence - they are for illustration of the concepts.
Biology
Serotonin is a ubiquitous neuromodulator with widespread effects, and one of these is to down-regulate GABAA (ionotropic chloride channel) receptors in projection pyramidal neurons of the pre-frontal cortex (PFC) in rats (Feng et al., 2001). In this system, serotonin activates the 5HT2 receptor subtype on the pyramidal neurons, which in turn activates intracellular protein kinase C, which in turn phosphorylates GABAA receptors. It is this last stage that actually down-regulates the GABA response. The PFC is involved in many higher-order cognitive functions, and GABA inhibition plays a crucial role in regulating its output, so serotonin modulation of that inhibition is also critically important. Dysfunction of this inhibition (and serotonin modulation in general) is thought to be a contributing factor in many neuropsychiatricSince serotonin is also present in large amounts in neurons controlling the gut, it is likely that serotonin malfunction has consequences beyond those involving the brain. disorders.
Pre-synaptic neuromodulation
There are many routes by which neuromodulators can regulate the release of transmitter from pre-synaptic neurons, but one established mechanism is to vary the duration of the pre-synaptic spike, and hence the amount of calcium inflow and the release of transmitter. The simplest way to achieve this is to modulate the conductance of the voltage-dependent potassium channels that contribute to the repolarization of the membrane after a spike.
- Load and Start the file Kv neuromod.
N1 (top trace, red) is a neuron that can generate Hodgkin-HuxleyThe kinetics are slightly modified from their canonical values.-type spikes. It receives 2 stimulus pulses, and generates 2 spikes. The conductance of its voltage-dependent potassium channel is shown (5th trace, green) since this is the property that is modulated in this model.
N1 makes an excitatory chemical synapse to N2 (2nd trace, blue).
N3 (3rd trace, orange) is not connected to the other two neurons directly, but it releases the neuromodulator serotonin (5HT) when it spikesThese spike are simple integrate-and-fire type, since their only job is to release the neuromodulator.. However, at this stage N3 is silent, and the extracellular 5HT concentration (bottom trace) remains at zero throughout.
- Click the up spin button for the amplitude of stimulus 2 (which is pre-selected in this simulation).
- A new simulation sweep occurs because the Run on change option is pre-selected.
N3 is now stimulated to spike just before the first N1 spike. It releases a bolus of 5HT causing a rise in its extracellular concentration. 5HT reduces the conductance of the voltage-dependent potassium channel in N1, which broadens the spike and increases its amplitude. Consequently the EPSP in N2 is enhanced. However, the concentration of 5HT declines fairly rapidly, and by the time of the second N1 spike it has almost reached its control (0) level. Hence the second EPSP in N2 is of approximately normal amplitude.
Note: As before (and as is often the case in these tutorials), this is a "concept" simulation designed to illustrate a key feature of a known mechanism. It is not intended to replicate the detailed physiology of a particular system.
Biology
One of the earliest studies of pre-synaptic neuromodulation was on the role of serotonin in sensitizing the withdrawal reflex in the marine mollusc AplysiaThis may sound like a rather obscure system to study, but it was the start of a series of investigations that eventually led to a Nobel prize for one of the principal investigators (Eric Kandel). (see Byrne & Kandel, 1996 for a review). In summary, a relatively benign touch stimulus to the tail, gill or siphon will usually elicit defensive withdrawal of the stimulated organ, and this reflex is largely mediated by monosynaptic excitatory connections between the relevant sensory and motor neurons. However, if the touch is preceded by a strongly noxious stimulus to an unrelated part of the body, the touch reflex is enhanced (sensitized). The noxious stimulus elicits release of serotonin from neuromodulator interneurons, and the serotonin acts on pre-synaptic sensory neurons to enhance their transmitter release. The first discovered mechanism of enhancement was down-regulation of an atypical potassium channel (Ks, where the s stands for serotonin) that was only weakly voltage-dependent. However, subsequent research revealed that serotonin also modulated the standard Kv channel, plus a variety of voltage-dependent calcium channels. Furthermore, serotonin also modulates the transmitter release mechanism directly, independently of the spike-broadening effect. However, spike broadening through down-regulation of voltage-dependent potassium channels is still thought to be a core component of the overall mechanism.
Voltage-Dependent Channel Neuromodulation
In the previous simulation, a neuromodulator down-regulated a voltage-dependent channel, but the end result (the "purpose") was to modulate synaptic transmission. However, in some cases, neuromodulation of a voltage-dependent channel can have profound effects on the properties of the neuron itself.
- Load and Start the file Conditional Plateau neuromod
N1 (top trace, blue) receives 3 brief (0.5 ms) stimulus pulses (2nd trace), and generates a single spike in response to each pulse. N2 (3rd trace, orange) releases the neuromodulator octopamine, but at this stage N2 is silent and the modulator concentration (bottom trace) remains at zero.
- Zoom in the Results view to see the second N1 spike more clearly by setting the Start time (left hand timebase) to 1000 ms and the End time (right hand timebase) to 1500 ms.
There is nothing very interesting about this spike (!).
- Double-click on N1 in the Setup view to open the Neuron Properties dialog.
Note that N1 has the two standardThe kinetics are modified from those in the squid axon, but the properties are basically similar. HH-type voltage-dependent channels (Na fast and K delayed rectifier), but that there is a third channel, Na persistent (Na-p). This is a voltage-dependent sodium channel, but it inactivates very slowly (hence its name, persistent). However, in control conditions the conductance of this channel is very low, so it has virtually no effect on the response to the stimulus (you can view its properties by selecting it in the list and checking the Channel properties box).
- Close the dialog.
- Click the up spin button for the amplitude of stimulus 2 (which is pre-selected in this simulation).
- The Results clear and a new simulation sweep occurs because the Auto clear and Run on change options are pre-selected.
Now two stimuli are applied to N2, after the first spike in N1. N2 spikes in response to the new stimuli, and the extracellular octopamine concentration rises following each N2 spike (bottom trace).
The elevated octopamine concentration up-regulates the conductance of the persistent sodium current in N1 (4th trace, green), which now so generates a long-lasting depolarization following the brief stimulus-induced initial spikeThere is a small depolarization before the stimulus due to octopamine causing a slightly elevated resting Na-p conductance.. This depolarization is known as a plateau potential, and it terminates when the persistent sodium channel eventually inactivates. N1 spikes repeatedly during this depolarization. The degree of up-regulation depends on the octopamine concentration, so the second plateau potential is stronger and longer-lasting than the first, because the octopamine concentration is higher.
- Again, change the timebase settings as above to zoom in the Results view to see the 2nd N2 response in more detail.
You can now clearly see the plateau potential, with the superimposed spikes, following the brief stimulus pulse.
- Check the octopamine box in the Drugs/Light frame of the Setup view.
Octopamine is now applied exogenously (at 1 unit concentration) throughout the experiment, so the first N1 stimulus elicits a plateau potential even though N2 is not active. The plateau potentials following the second and third stimuli are significantly enhanced, as the exogenous octopamine effect sums with that of the N2-released octopamine.
- Check the PRAX-562 box in the Drugs/Light frame.
This drug blocks persistent sodium channels (Kahlig et al., 2022), and hence prevents the induction of plateau potentials in this model. N1 now responds to each stimulus with a single spike, just as it did in the absence of octopamine.
Biology
Octopamine is a major neuromodulator in invertebratesIt was first identified in the salivary glands of the octopus, hence its name.. It is chemically related to noradrenaline (norepinephrine) found in vertebrates, and often seems to perform a similar "flight-or-fight" function in preparing the animal for action. It has been shown to activate plateau properties in certain insect neurons (Ramirez & Pearson, 1991), although the functional/behavioural significance of this is unclear. The mechanism underlying the octopamine-induced production of plateau potentials is not known, but enabling a persistent sodium current is a plausible, although completely speculative, hypothesis.
Dendritic Computation
An individual neuron may receive hundreds or thousands of synaptic inputs from different pre-synaptic neurons, and it is the interplay between these inputs, and the way they are integrated, that forms the basis of decision making in the nervous system. If the summed synaptic inputs take a spiking neuron above threshold, then a spike is generated and a signal passes down its axon and on to the next neuron. If threshold is not crossed, no spike occurs, and no signal is passed on. If the neuron operates with non-spiking synapses, then the summed potential is the signal, and no spikes are necessary.
Many synaptic inputs occur on the dendrites of neurons, and these can have very complex branching structures. Where and when an input occurs on this structure can have important consequences for signal processing.
Proximal and Distal Inhibition
- Load the parameter file Proximal Distal Inhibition, but do not Start it yet.
The Setup view shows a highly simplified compartmental model of a vertebrate motorneuron. At the top there is a cell body (soma; N1), which has just one dendrite sprouting off it (N2-7; the vertical line of violet rectangles). The dendrite is non-spiking, but the cell body generates integrate-and-fire spikes if its membrane potential goes above threshold. For simplicity and speed the model does not include an axon, but if the cell body spiked, those spikes would propagate down the axon.
The distal dendritic terminal (N7; bottom) receives an excitatory synaptic input (type a) from a pre-synaptic neuron (N8; green). It also receives distal inhibitory synaptic input (type b) from another pre-synaptic neuron (N9; pink). There is another inhibitory synaptic input (type c) from yet another pre-synaptic neuron (N10, magenta), but this occurs on the proximal dendrite, just before it enters the cell body.
Once you have identified the various components in the Setup view
- Select the Neuron: Colours: Colour from voltage menu command.
Now the elements in the Setup view are coloured-coded by their membrane potential. At the moment all are at rest, hence their dark blue colour.
- Click Start.
The distal pre-synaptic excitatory neuron is stimulated (stimulus 1) and spikes, delivering a large EPSP to the distal dendrite. This propagates through passive conduction to the soma. The resulting depolarization is above threshold, so the soma spikes.
- Change the duration of stimulus 1 to 55 ms.
- Run on change and Auto clear are pre-selected, so the Results clear and a new sweep is initiated as soon as you make the change.
The longer duration stimulus causes a burst of spikes in the pre-synaptic excitatory neuron. The EPSPs in the distal dendrite summate. This simply extends the response so that at the end of the sweep you can still see the colour changes in the Setup view reflecting the membrane potential of the elements.
- Select stimulus 2.
- Click the up spin button of the stimulus Amplitude to change the value from 0 to 1 nA.
This activates the distal pre-synaptic inhibitory neuron, which now spikes. The resulting IPSPs negatively summate with the distal EPSPs and pull the membrane potential down somewhat. The soma no longer spikes.
We now want to edit the synaptic properties.
- Select the Synapses: Spiking chemical menu command to show the Spiking Chemical Synapse Types dialog box.
- There is a list of synapse types at the top of the dialog box. We are only interested in types a, b and c. You can see the properties of these in the main body of the dialog by clicking them.
- Note the equilibrium potential and strength of each type of these synapses. The strength is given by the Synaptic conductance parameter.
- The dialog is non-modal and can be kept open while the simulations run. However, it is best to move it to a location where it does not obscure the main program.
You are now going to make the distal EPSP stronger, to see if it can overcome the inhibition.
- Make sure that synapse Type a is selected (EPSP distal).
- Click the up spin button of the Synaptic conductance parameter, to change it from 4 to 8 mS/cm2.
- Note: This produces exactly the same effect as recruiting an identical second excitatory pre-synaptic neuron would. In a real system it would not necessarily be easy to double the strength of an existing synapse, but it would presumably be easy to recruit another existing pre-synaptic excitatory neuron.
The stronger distal EPSP counteracts the distal IPSP, and once again the soma spikes.
- Select synapse Type b (IPSP distal) in the Synapse dialog.
- Click the up spin button of the Synaptic conductance parameter, to change it from 2 to 4 mS/cm2.
The additional inhibition does not completely block the soma spikes, but it reduces their frequency.
- Repeatedly click the up spin button of the inhibitory synapse conductance until the soma stops spiking.
- Switch back to the excitatory synapse and repeatedly click the up spin button of the conductance until the soma spikes.
- Repeat a few times until you are convinced that you can always overcome distal excitation by increasing distal inhibition, and you can always overcome distal inhibition by increasing distal excitation.
What is going on? Essentially, the membrane potential of the distal compartment of the dendrite is a weighted compromise between the equilibrium potentials of the EPSPs (-10 mV) and IPSPs (-80 mV), with the weighting determined by their relative conductancesThis is like the Goldman equation described in the Passive Properties tutorial.. So you can drive the membrane potential closer to -10 mV by increasing excitatory conductance, and closer to -80 mV by increasing inhibitory conductance.
- Click Revert in the Synapse dialog to put the conductances of both types back to their default values.
- Reset stimulus 2 to 0 to turn off the distal pre-synaptic inhibitory neuron (the soma should now spike again).
- Select stimulus 3.
- Click the up spin button of the Amplitude parameter to set it to 1 nA.
You are now activating the proximal pre-synaptic inhibitory neuron. This synapse (Type c; IPSP proximal) is relatively weak (its synaptic conductance is only 0.5 mS/cm2), but it still prevents spikes in the soma. Can this inhibition be overcome by increasing distal excitation?
- Select synapse Type a (EPSP distal) in the Synapse dialog.
- Hand edit the synaptic conductance to 100 mS/cm2.
You have produced a massive increase in distal excitation, but this has hardly any effect at the soma, which still fails to spike. The massive distal excitation cannot overcome the weak proximal inhibition.
Question: Why cannot the proximal inhibition be overcome by increasing distal excitation?
I’m going to try to explain this (based on Vu and Krasne, 1992), so if you are stuck on the question, just read on. However, you might find it useful to try to answer it yourself before reading the next section.
When an EPSP occurs on the distal dendrite, intracellular current flows from the distal to the proximal region because the distal membrane is depolarized relative to the proximal membrane. The greater that depolarization, the more current flows (Ohm's Law) and the greater the excitatory effect. However, there is a ceiling to the voltage that the distal dendrite can ever reach, and that is the equilibrium potential of the excitatory synapse (-10 mV in this case). So a massive increase in distal excitatory synaptic conductance “locks” the distal dendrite firmly to that voltage, but can never take it beyond it – it cannot make it still more positive. Distal inhibition will tend to drag the distal membrane potential negative from this equilibrium potential, so increasing excitatory conductance can pull it back up again and restore excitation – the two can compete with each other.
Proximal inhibition does not work by pulling the distal membrane potential negative. It simply tries to lock the membrane potential at the proximal site to the inhibitory equilibrium potential. Since the excitatory synapse cannot increase the distal voltage above its own equilibrium potential, it cannot drive more positive current proximally to overcome the proximal inhibition, no matter how strong it is.
As a confirmation of this hypothesis:
- With synapse Type a (EPSP distal selected) change the Equilibrium potential from -10 to +100 mV.
- This is a completely unrealistic value for a normal EPSP, but it tests the theory.
The soma now spikes!
It is likely that evolution tunes the location of inhibition according to its function. If responding to an excitatory stimulus would be seriously inappropriate (e.g. fatal) in a particular situation, that situation should activate proximal inhibition, with the excitation being delivered distally. The inhibition can then override the excitation, no matter how strong the latter is. But if the inhibition and excitation should compete on more-or-less equal terms, as in finely-balanced decision making, then they should be co-located in the same distal dendritic region, so that the outcome just depends on which is the stronger.
Take-home message. Distal inhibition can be overcome by increasing distal excitation, but proximal inhibition can be absolute – if it is above a certain strength, it cannot be overcome by increasing distal excitation.
Directional Sensitivity
- Load the parameter file Directional Dendrite.
The Setup view shows two identical small circuits, labelled Circuit 1 and Circuit 2. The elements within each circuit are colour-coded according to their membrane potential. Initially, all are at rest and have a dark blue colour.
Within each circuit, on the left is a simplified compartmental model of a sensory interneuron. As in the previous simulation, there is just one dendrite (the vertical line of rectangles), which connects to the cell body at the top (the purple circle). The dendrite is non-spiking, but the cell body generates integrate-and-fire spikes if its membrane potential goes above threshold. For simplicity and speed the circuit does not include an axon, but if the cell body spiked, those spikes would propagate along the axon.
On the right within the circuit is an array of sensory neurons which make mapped input onto the dendrite. These are supposed to have contiguous receptive fields in real space. The synaptic inputs are all identical.
Each sensory neuron within a circuit is activated in turn by a stimulus, and the two circuits get activated simultaneously. The only difference between the two circuits is the order in which the sensory neurons get activated.
- Look at the Circuit 1 in the Setup view and click Start.
- You may need to set a Slow down factor in the main toolbar.
The stimuli have been arranged so that each sensory neuron is activated in a bottom-to-top temporal sequence - you can see their colour change as they are activated. This is the equivalent to a sensory stimulus sweeping across the receptive field from bottom to top . The dendrite thus receives excitatory synaptic input in a distal-to-proximal temporal sequence.
In the Results view, the top axis shows all the sensory neuron responses in circuit 1 superimposed (shades of blue). The second axis shows the interneuron membrane potential at the soma (the red trace). The EPSPs impinging on the dendrite passively conduct to the soma, where they (just) summate sufficiently to cause a spike.
- Look at the Circuit 2 in the Setup view and click Start.
In circuit 2 the colour change sweeps from top to bottom, which is as though the sensory stimulus swept across the receptive field in the opposite direction (i.e. proximal-to-distal). In the Results view it is apparent that the sensory neurons in the third axis are activated in the opposite sequence. There is less summation at the soma, and the soma does not spike.
It is clear that soma 1 shows a progressive and approximately linear depolarization that eventually reaches threshold, while soma 2 shows a more rapid early depolarization, but this then plateaus before it reaches threshold. The exact quantitative reason for this difference is not (at least to me) immediately obvious, so this is a case where simulation can test something that could not be predicted with confidence in advance.
We all know from subjective experience that we can detect the difference between seeing something moving downwards in our field of view, and the same object moving upwards. What this simulation shows is that the difference could in theory be detected even at the level of a single neuron (although of course there is a lot more than that going on in our brains).
Take-home message: Synaptic integration can depend both on the spatial arrangement of synaptic inputs on the dendritic tree, and on the temporal sequence in which they are activated.
On to Networks ...